Los síntomas asociados a la PV son frecuentes y clínicamente significativos. De forma general, se distinguen síntomas constitucionales o generales de las NMPs, síntomas microvasculares y complicaciones macrovasculares.
En el momento del diagnóstico, la carga sintomática en pacientes con PV es comparable a la de pacientes con MFP o mieloma múltiple.
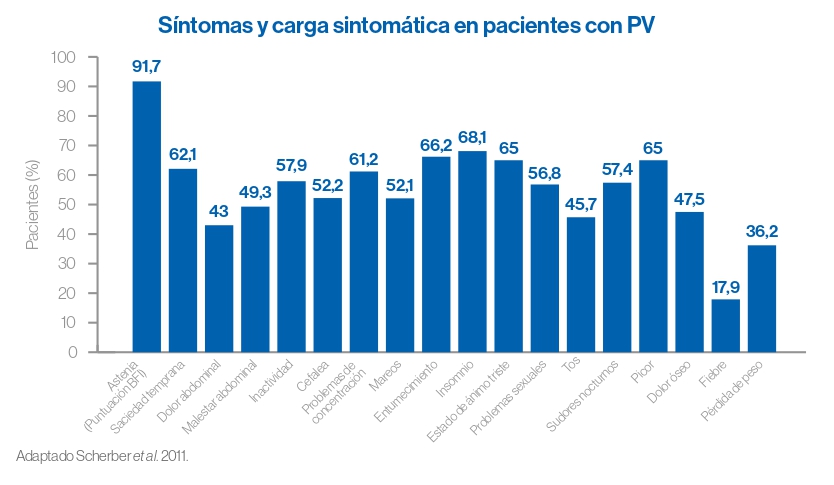
Los principales síntomas de PV están relacionados con anomalías provocadas por la elevada masa de eritrocitos, aunque pueden ocurrir independientemente de los valores sanguíneos, duración de la enfermedad y el tratamiento mielosupresor.
La aparición de prurito y astenia se encuentran entre los síntomas más habituales y graves de la PV. El prurito, especialmente después de un baño o ducha, se atribuye a hipersensibilidad a la degranulación de mastocitos y basófilos, con la liberación de histamina, factores fibrinolíticos, prostaglandinas o interleuquina-3.
Algunos pacientes con PV reportan síntomas constitucionales como: dolor de cabeza, debilidad, fiebre, molestias abdominales, sudoración nocturna, dolor óseo, saciedad temprana, inactividad, caquexia y problemas de concentración.
Estos síntomas asociados son provocados por un incremento en la producción de citoquinas inflamatorias derivado de la activación anómala de la vía JAK-STAT.
Los síntomas gastrointestinales, como dispepsia no ulcerosa por úlcera gástrica, también son comunes en PV.
La escala de síntomas validada MPN-10 es una herramienta útil para evaluar la evolución de los síntomas en el paciente de PV, calculando una suma total en base a la puntuación que el paciente ha asignado a cada síntoma individual.
En un estudio reciente, la reducción de síntomas fue el objetivo más importante para los pacientes de PV. Siendo la fatiga/cansancio los síntomas que más querían remediar.
Las complicaciones trombohemorrágicas son la principal causa de morbilidad y mortalidad en PV en su historia natural.
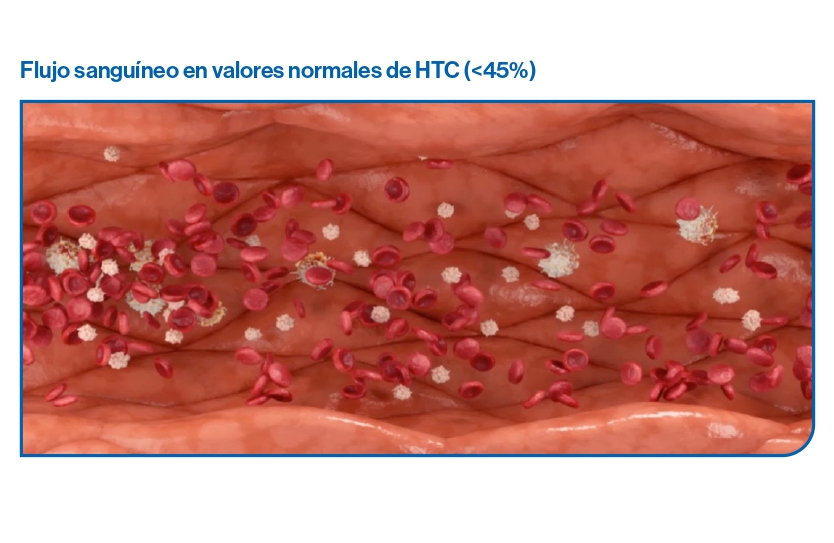
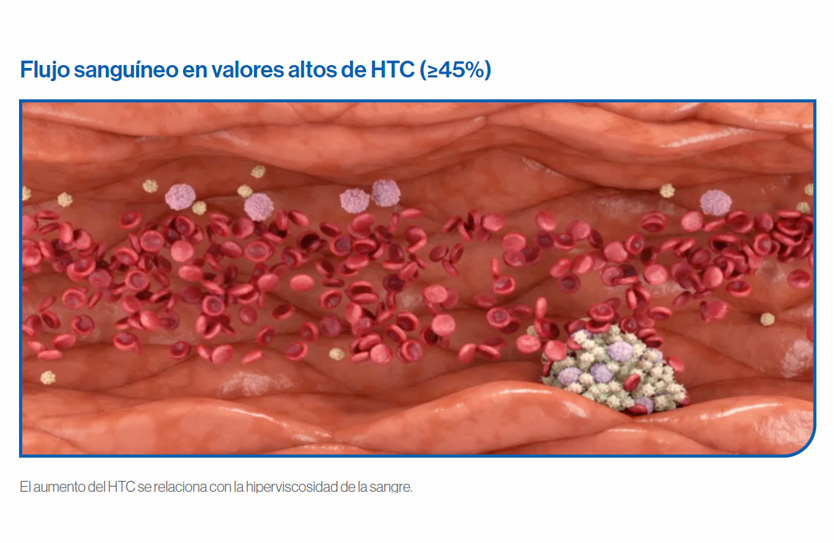
La causa de la trombosis en PV es la producción anómala de células sanguíneas que resulta en un incremento de la viscosidad sanguínea, junto con las anomalías cuantitativas y cualitativas observadas a nivel de plaquetas y leucocitos y el estado inflamatorio.
El aumento de la viscosidad de la sangre en casos con hematocritos más elevados y un volumen sanguíneo en el rango normal, altera el flujo sanguíneo y reduce el transporte de oxígeno.
El aumento del volumen sanguíneo por eritrocitosis amplía el lecho vascular, disminuye la resistencia periférica y aumenta el gasto cardíaco. Además, provoca que el flujo sanguíneo sea axial, con un flujo central de glóbulos rojos circulantes que se desliza sobre una capa periférica de plasma lubricante.
La mutación de JAK2 produce una sobreestimulación de la vía, lo que conduce a un aumento de la proliferación celular. Esta mutación provoca que la estimulación de la vía sea independiente de la unión de los factores a su receptor y por tanto esté activa en presencia o ausencia de los ligandos. La consecuencia de esta hiperestimulación, entre otras, es la hipercoagulabilidad, provocada por hiperviscosidad celular, cambios en los factores del endotelio y en la adhesión y función celular.
Referencias:
1. Levine RL, et al. Nat Rev Cancer. 2007 Sep;7(9):673-83. 2. Lodish H, et al. Molecular Cell Biology. 6th ed. New York, NY: W. H. Freeman; 2008. 3. Vardiman JW, et al. Blood. 2009 Jul 30;114(5):937-51. 4. Stuart BJ, Viera AJ. Am Fam Physician. 2004 May 1;69(9):2139-44. 5. Hensley B, et al. Expert Opin Pharmacother. 2013 Apr;14(5):609-17. 6. Swerdlow SH, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2008. 7. Spivak J. N Engl J Med. 2004 Jan 8;350(2):99-101. 8. Falanga A, et al. Blood. 2000;96:7. 9. Alvarez-Larrán A, et al. Br J Haematol. 2004;124:329-35. 10. Arellano-Rodrigo E, et al. Haematologica. 2006;169:75. 11. Falanga A, et al. Exp Hematol. 2007;35:702–11. 12. Alvarez-Larrán A, et al. Ann Hematol. 2008;87:269–76. 13. Torregrosa JM, et al. Br J Haematol. 2016 Mar;172(5):813-5. 14. Guy A, et al. Leukemia. 2019;33:2544–8. 15. Jensen MK, et al. Br J Haematol. 2000;110:116–24. 16. Arellano‐Rodrigo E, et al. Am J Hematol. 2008;84:102–8. 17. Charpentier A, et al. Haematologica. 2016;101:e365–8. 18. Trappenburg MC, et al. Haematologica. 2009;94:911–8. 19. Tong D, et al. Ann Hematol. 2018;97:605–16. 20. Barbui T, Falanga A. Thromb Res. 2016 Apr;140 Suppl 1:S71-5. 21. Falanga A, Marchetti M. Hematology Am Soc Hematol Educ Program. 2012;2012:571-81. 22. Zawrotniak M, Rapala-Kozik M. Acta Biochim Pol. 2013;60(3):277-84. Epub 2013 Jul 1. 23. Gordeuk VR, et al. Haematologica. 2019 Apr;104(4):653-658. 24. Lodish H et al. Molecular Cell Biology. 6th ed. New York, NY: W. H. Freeman; 2008. 25. Levine RL, et al. Nat Rev Cancer. 2007 Sep;7(9):673-83. 26. Perner F, et al. Cells. 2019 Aug 8;8(8):854.
La esplenomegalia está presente de un 35%-45% de los pacientes con PV. Numerosos síntomas clínicos derivan de la esplenomegalia, incluyendo dolor abdominal, saciedad temprana, disnea, tos o infarto esplénico.
Es de destacar que la esplenomegalia se ha relacionado con la PV no controlada, debido a su asociación a un mayor riesgo a transformación a MF.
Algunos pacientes sufren síntomas derivados de alteraciones microcirculatorias, que son causadas por la obstrucción de la circulación sanguínea a través de vasos sanguíneos pequeños. Estos síntomas incluyen eritromelalgia, dolor de cabeza, mareos, alteraciones visuales y auditivas, fenómenos de tipo Raynaud o tromboflebitis superficial, parestesia y accidente isquémico transitorio.
La transformación en leucemia aguda mieloblástica o síndromes mielodisplásicos (LAM/SMD) es una de las posibles evoluciones de la PV. Sin embargo, es más probable la transformación a MF secundaria. Después de más de 10 años de tratamiento, un 10% de pacientes desarrollan MF secundaria con esplenomegalia y citopenia, mientras que hasta un 6% de pacientes evolucionan a LAM.
Referencias:
1. Swerdlow SH, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2008. 2. Chou YS, et al. Eur J Haematol. 2013;90:228-236. 3. Tefferi A, et al. Leukemia. 2013;27:1874-1881. 4. Luo EJ, Levitt L. Hosp Physician. 2008;44:3. 5. Bhat, Vivek, et al. Journal of Stroke and Cerebrovascular Diseases 31.1 (2022): 106167. 6. Levine RL, et al. Nat Rev Cancer. 2007;7:673-83. 7. Lodish H, et al. Molecular Cell Biology. 6th ed. New York, NY, USA: W. H. Freeman & Co Ltd; 2008. 8. McMullin MF, et al. Br J Haematol. 2016;172(3):337-49.
Referencias:
1. Abelsson J, et al. Leuk Lymphoma. 2013;54:2226-2230.
2. Scherber R, et al. Blood. 2011;118:401-408.
3. Stein BL, et al. Ann Hematol. 2014;93(12):1965-76.
4. Smith CA, Fan G. Hum Pathol. 2008;39:795-810.