La mielofibrosis (MF) es una neoplasia mieloproliferativa crónica cromosoma Filadelfia negativa potencialmente mortal asociada a una desregulación de la vía de señalización JAK-STAT.
Se origina por la transformación de una célula madre hematopoyética, que da lugar a mieloproliferación crónica e hiperplasia megacariocítica atípica, seguidas de cambios estromales que desencadenan la aparición de la fibrosis característica en el tejido medular.
La mielofibrosis (MF) puede aparecer de novo (MF primaria; MFP) o por evolución de alguna de las otras dos neoplasias mieloproliferativas crónicas Filadelfia-negativas clásicas: MF secundaria a policitemia vera (MF post-PV) o secundaria a trombocitemia esencial (MF post-TE).
Aproximadamente un 15% de pacientes con PV o TE progresan a MF. La MF ocasiona principalmente anemia, esplenomegalia y toda una variedad síntomas progresivos y debilitantes causados tanto por la mieloproliferación como por el aumento de los niveles de diferentes citoquinas inflamatorias.
Hasta el desarrollo clínico de Jakavi®, las terapias utilizadas de forma convencional para el tratamiento de la MF habían sido principalmente de tipo sintomático, y ninguno de los tratamientos farmacológicos existentes había sido evaluado en ensayos clínicos controlados y aleatorizados.
Si bien el trasplante alogénico de células progenitoras hematopoyéticas, es una opción potencialmente curativa, en torno al 90% de los pacientes de MF no son candidatos debido a los riesgos asociados que presenta.
A nivel molecular, la mielofibrosis está asociada a la desregulación de la transducción de señales de la vía JAK-STAT. Las mutaciones principales afectan a JAK2 (50-60%), calreticulina (20-30%) y MPL (5-10%). Alrededor del 10% de los pacientes no presentan mutaciones en ninguno de estos genes driver, lo que se conoce como casos “triple negativo”. Cada vez más, se conocen otras mutaciones accesorias con diferente impacto sobre la evolución de la enfermedad.
Referencias:
1. Arber DA, et al. Blood. 2016;127(20):2391-405. 2. Mascarenhas J, Hoffman R. Clin Cancer Res. 2012;18(11):3008-14. 3. Tefferi A, et al. Leukemia. 2008;22: 14-22. 4. Manual recomendaciones GEMFIN 2020. 5. Barosi et al. Leukemia, 2008 Volume 22, pages 437–438. 6. Baumeister, J. et al. Cells, 2021, 10, 3551. 7. Harrison C, Vannucchi A. Blood, 2016;127:276-278
La mielofibrosis es una enfermedad poco frecuente, se estima que la incidencia anual de la MF es de entre 0,1 y 1 caso por cada 100.000 habitantes y año.

Esta baja incidencia y la relevancia clínica hace que la MF se categorice como una enfermedad huérfana.
Si bien la MF afecta mayoritariamente a personas de edad avanzada, esta enfermedad acorta la supervivencia de los pacientes, tal como se ha comprobado al compararla con la de individuos control. Con todo, dicha supervivencia ha ido aumentando a lo largo del tiempo y en la actualidad la mediana se acerca a los 7 años. No obstante, existe una gran heterogeneidad, ya que algunos pacientes fallecen en uno o dos años mientras otros viven más de 20 años. Las principales causas de muerte en la MF son la evolución a leucemia aguda (alrededor del 20%), la progresión de la enfermedad con caquexia e intenso debilitamiento, la infección, la hemorragia, la hipertensión portal, las trombosis en otros territorios y causas no relacionadas con la MF, en especial segundas Neoplasias.
Referencias:
1. Manual de recomendaciones NMP Grupo Gemfin 2020.
2. Pastor-Galan et al, Medicina clínica 2020.
La MF es un trastorno clonal de la hematopoyesis, si bien las causas exactas de la regulación anómala inicial de las células madre aún son objeto de estudio y discusión.
En la MF se pueden considerar dos mecanismos patogénicos interdependientes:
- El proceso patológico primario en la MF (fase celular) es un trastorno clonal de una célula madre hematopoyética que se traduce en mieloproliferación crónica e hiperplasia de megacariocitos atípicos.
- El componente secundario del proceso patogénico son los cambios estromales de la médula ósea, que incluyen fibrosis, osteosclerosis, angiogénesis y hematopoyesis extramedular (en el bazo, el hígado y otros lugares).
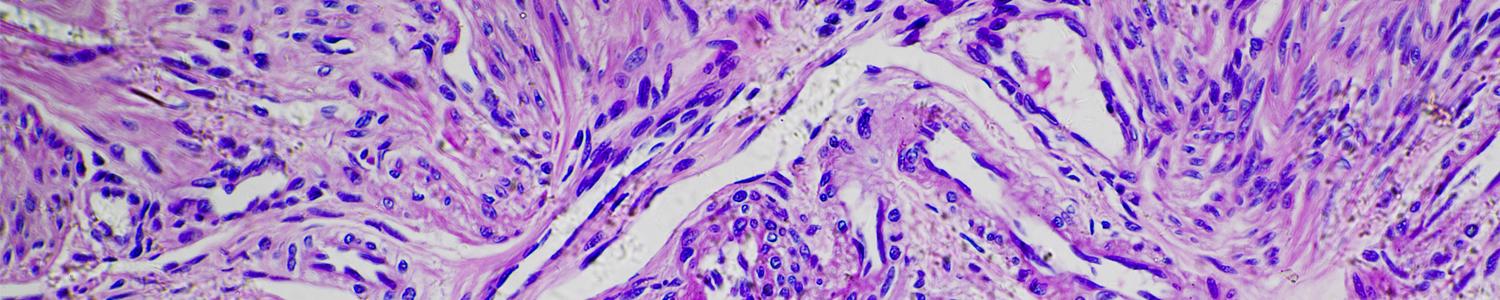
La principal característica de la MF es el desarrollo de fibrosis en la médula, producida por una gran deposición de colágeno por los fibroblastos no neoplásicos.
Referencias:
1. Levine RL et al. Nat Rev Cancer 2007.
2. Manual de recomendaciones GEMFIN 2020.
3. Vainchenker W, Kralovics R. Blood. 2017;129(6):667–79.
La MF está asociada a la desregulación de la transducción de señales de la vía JAK-STAT. Se cree que la base de esta desregulación se relaciona con los siguientes factores:
Mutaciones de ganancia de función
La mutación JAK2V617F se produce en aproximadamente la mitad de los pacientes con MF o TE, y en la mayoría de pacientes con PV. La proteína mutante está activada constitutivamente, lo que provoca una activación continua de transmisores de señales y activadores de la transcripción (STAT. Signal transducers and Activators of transcription).
Las mutaciones del receptor de trombopoyetina (mutación MPLW515L/K) también son capaces de activar la señalización JAK-STAT en pacientes con MF.
Por otro lado, se han identificado mutaciones en el gen de la calreticulina o CALR (una chaperona asociada al retículo endoplásmico) en la mayoría de pacientes con MFP que no presentan mutaciones en JAK2 o MPL. Se ha sugerido que estas mutaciones también son capaces de hiperactivar la señalización de la vía JAK-STAT.
En los últimos años, un mayor uso de las técnicas de secuenciación masiva ha contribuido a la identificación de otras mutaciones no directamente relacionadas con la vía JAK-STAT, pero con un papel importante en la mielofibrosis como es el caso de TET2, ASXLl, EZH2, IDHl/2, CBL, que afectan a genes implicados en la regulación de la expresión génica y tienen interés pronóstico.
Niveles altos de citoquinas circulantes que activan la vía JAK-STAT
Las proteínas JAK ejercen un papel muy importante en la transmisión de la señalización de citoquinas infamatorias.
La mayoría de pacientes con MFP presentan niveles elevados de citoquinas pro-inflamatorias que ejercen su efecto a través de la vía JAK-STAT (por ejemplo IL-6).
La inflamación derivada del efecto de las citoquinas pro-inflamatorias puede dirigir la expansión clonal de la enfermedad de un estadio temprano a estadios más avanzados. La citoquina proinflamatoria TNFα es capaz de facilitar la expansión clonal de células JAK2V617F positivas. Las interleuquinas IL-8, IL-12 e IL-15 y el receptor de IL-2 son indicadores pronósticos de menor supervivencia y transformación a leucemia en pacientes con MFP. Las citoquinas son también responsables de muchos síntomas que presentan los pacientes con MF.
Silenciamiento de los mecanismos reguladores negativos de la vía JAK-STAT
Se han identificado mutaciones en la proteína adaptadora LNK en pacientes con NMP sin la mutación JAK2V617F o mutaciones en el gen MPL. Estos pacientes presentan perdida de la función inhibitoria de LNK sobre JAK2 y un incremento en la señalización de JAK-STAT.
Se ha observado que los pacientes con MF denominada "triple negativa" (sin la mutación JAK2V617F, o mutaciones en el gen CALR ni MPL) parecen tener un peor pronóstico.
En cualquier caso, los pacientes con MF presentan desregulación de la transducción de señales de la vía JAK-STAT, independientemente de la mutación existente.
Referencias:
1. Vainchenker W, Kralovics R. Blood. 2017;129(6):667–79. 2. Harrison C, Vannucchi A. Blood 2016;127:276-278. 3. Levine RL, et al. Nat Rev Cancer. 2007;7:673-83. 4. Grinfeld et al. N Engl J Med 2018;379:1416-30. 5. Vannucchi AM, et al. Leukemia. 2013;27:1861–9.

